A special aspect of X-ray spectroscopy calculations which involve an explicit core-hole, e.g. X-ray photoelectron spectroscopy (XPS) or X-ray emission spectroscopy (XES), is the delocalization-induced relaxation error (DIRE) Brumboiu & Fransson (2022). DIRE affects symmetric molecules where care must be taken to localize the core-hole, especially when HF or DFT theory levels are used. Below follows an illustration of DIRE in the case of the ethylene molecule. We will calculate the ionization energy of the C 1s core orbitals. The ionization energy is calculated as:
We will have to perform a ground state calculation, as well as a calculation with a full core-hole (FCH) on the C 1s orbital. The FCH is obtained using the maximum overlap method (MOM). One option to localize the core-hole is to replace the 1s electrons of the non-core-ionized C atom with an effective core potential (ECP). Another option is to very slightly distort the molecule, which is the approach adopted here.
Step 1 Define the molecular geometries¶
In the following, we will create two ethylene molecules, one fully symmetric to obtain delocalized C 1s core orbitals and and one with slightly stretched CH bonds at one of the C sites, to obtain localized C 1s core orbitals.
deloc_ethylene_xyz = """6
symmetric ehtylene molecule
C 0.666005 0.000000 0.000000
C -0.666005 0.000000 0.000000
H 1.227874 0.922836 0.000000
H 1.227874 -0.922836 0.000000
H -1.227874 0.922836 0.000000
H -1.227874 -0.922836 0.000000
"""
loc_ethylene_xyz = """6
asymmetric ethylene molecule to localize the C1s orbitals
C 0.666005 0.000000 0.000000
C -0.666005 0.000000 0.000000
H 1.227874 0.922836 0.000000
H 1.227874 -0.922836 0.000000
H -1.2538762 0.965543 0.000000
H -1.2538762 -0.965543 0.000000
"""
basis_set_label = "6-311++G_D,P_" #"DEF2-TZVPPD" #"TAUG-CC-PCVDZ" #
deloc_ethylene = vlx.Molecule.from_xyz_string(deloc_ethylene_xyz)
loc_ethylene = vlx.Molecule.from_xyz_string(loc_ethylene_xyz)
basis = vlx.MolecularBasis.read(loc_ethylene, basis_set_label)Step 2 determine the ground state energy¶
deloc_scf_drv = vlx.ScfRestrictedDriver()
deloc_scf_drv.ostream.mute()
deloc_scf_results = deloc_scf_drv.compute(deloc_ethylene, basis)loc_scf_drv = vlx.ScfRestrictedDriver()
loc_scf_drv.ostream.mute()
loc_scf_results = loc_scf_drv.compute(loc_ethylene, basis)plot_orbital(deloc_ethylene, basis, deloc_scf_drv, orbital_index=0)plot_orbital(loc_ethylene, basis, loc_scf_drv, orbital_index=0)Step 3 determine the core-ionized state energy¶
def compute_fch_energy(molecule, basis, scf_drv):
fch_molecule = copy.deepcopy(molecule)
fch_molecule.set_charge(1)
fch_molecule.set_multiplicity(2)
# Lists of occupied alpha and beta MOs
occ_alpha = [0, 1, 2, 3, 4, 5, 6, 7]
occ_beta = [1, 2, 3, 4, 5, 6, 7]
# Calculate SCF of the core-ionized system using the maximum overlap method
scfdrv_fch = vlx.ScfUnrestrictedDriver()
scfdrv_fch.ostream.mute()
scfdrv_fch.maximum_overlap(fch_molecule, basis, scf_drv.mol_orbs,
occ_alpha, occ_beta)
scf_results = scfdrv_fch.compute(fch_molecule, basis)
return scfdrv_fch.get_scf_energy()deloc_fch_energy = compute_fch_energy(deloc_ethylene, basis, deloc_scf_drv)
deloc_gs_energy = deloc_scf_drv.get_scf_energy()
deloc_hf_ie = (deloc_fch_energy - deloc_gs_energy) * hartree_in_ev()
# Add a Lorentzian broadening
deloc_x, deloc_y = lorentzian(np.array([deloc_hf_ie]), np.array([1]),
deloc_hf_ie - 5, deloc_hf_ie + 5, 0.01, 0.7)loc_fch_energy = compute_fch_energy(loc_ethylene, basis, loc_scf_drv)
loc_gs_energy = loc_scf_drv.get_scf_energy()
loc_hf_ie = (loc_fch_energy - loc_gs_energy) * hartree_in_ev()
loc_x, loc_y = lorentzian(np.array([loc_hf_ie]), np.array([1]),
loc_hf_ie - 5, loc_hf_ie + 5, 0.01, 0.7)Now that we have calculated the ionization energies for both the localized and delocalized CH, let us plot the results. The dotted line marks the measured ionization energy Holme et al. (2011).
plt.figure(figsize=(7, 4))
plt.bar(deloc_hf_ie, 1.0, color="darkred", width=0.1)
plt.bar(loc_hf_ie, 1.0, color="navy", width=0.1)
plt.plot(deloc_x, deloc_y, color="darkred", label="deloc. CH", linewidth=2.5)
plt.plot(loc_x, loc_y, color="navy", label="loc. CH", linewidth=2.5)
plt.xlabel("Core-ionization energy (eV)")
plt.ylabel("Intensity (arb. units)")
plt.title("Ethylene, HF/6-311++G(d,p)")
plt.axvline(290.7, linestyle="--", color="gray", label="exp.")
plt.axis(xmin=287.5, xmax=300.5, ymin=-0.1, ymax=4.0)
plt.xticks([288, 290, 292, 294, 296, 298, 300])
plt.yticks([0])
plt.legend()
plt.show()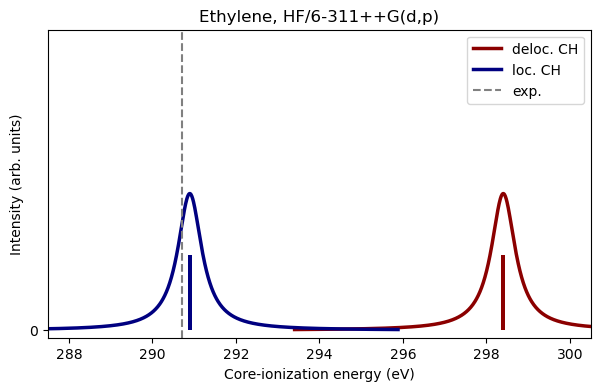
Source
print("DIRE, HF XPS: %.2f eV" % (deloc_hf_ie - loc_hf_ie) )DIRE, HF XPS: 7.51 eV
- Brumboiu, I. E., & Fransson, T. (2022). Core-hole delocalization for modeling x-ray spectroscopies: A cautionary tale. J. Chem. Phys., 156, 214109. 10.1063/5.0088195
- Holme, A., Børve, K. J., Sæthre, L. J., & Thomas, T. D. (2011). Accuracy of Calculated Chemical Shifts in Carbon 1s Ionization Energies from Single-Reference ab Initio Methods and Density Functional Theory. J. Chem. Theory Comput., 7, 4104. 10.1021/ct200662e