import veloxchem as vlxDefine the molecule¶
We first define the structure of a water molecule and choose a basis set.
mol_xyz = """3
O 0.0000000000 0.1178336003 0.0000000000
H -0.7595754146 -0.4713344012 -0.0000000000
H 0.7595754146 -0.4713344012 0.0000000000
"""
molecule = vlx.Molecule.read_xyz_string(mol_xyz)
basis = vlx.MolecularBasis.read(molecule, "cc-pVDZ", ostream=None)molecule.show()Loading...
print("Number of atoms:", molecule.number_of_atoms())
print("Number of electrons:", molecule.number_of_electrons())
print("Number of contracted basis functions:", basis.get_dimensions_of_basis())Number of atoms: 3
Number of electrons: 10
Number of contracted basis functions: 24
SCF optimization¶
Perform a self-consistent field (SCF) optimization to obtain the Hartree–Fock wave function and the associated ground-state energy.
scf_drv = vlx.ScfRestrictedDriver()
scf_drv.ostream.mute()
scf_results = scf_drv.compute(molecule, basis)SCF information¶
The SCF driver object has a method named get_scf_energy() for retrieving the final energy.
print(f"Hartree–Fock energy: {scf_drv.get_scf_energy():14.10f} a.u.")Hartree–Fock energy: -76.0265782198 a.u.
The return object from the compute() method is a Python dictionary containing several tensors:
C: molecular orbital coefficients as a NumPy arrayE: orbital energies as a NumPy arrayD: - and -spin density matrices as a tuple of NumPy arraysF: - and -spin Fock matrices as a tuple of NumPy arraysS: overlap integrals as a NumPy array
print("Dictionary keys:\n", scf_results.keys())
print("\nOrbital energies:\n", scf_results["E_alpha"])Dictionary keys:
dict_keys(['eri_thresh', 'scf_type', 'scf_energy', 'restart', 'filename', 'S', 'C_alpha', 'C_beta', 'E_alpha', 'E_beta', 'occ_alpha', 'occ_beta', 'D_alpha', 'D_beta', 'F_alpha', 'F_beta', 'F', 'dipole_moment'])
Orbital energies:
[-20.55119961 -1.33473346 -0.6969265 -0.56605275 -0.49289826
0.18486487 0.25566978 0.7860471 0.85103488 1.16387193
1.20030818 1.25366154 1.44422794 1.4755769 1.67329727
1.8679223 1.93111284 2.4430781 2.48048697 3.28268216
3.334531 3.50569215 3.86065795 4.14355138]
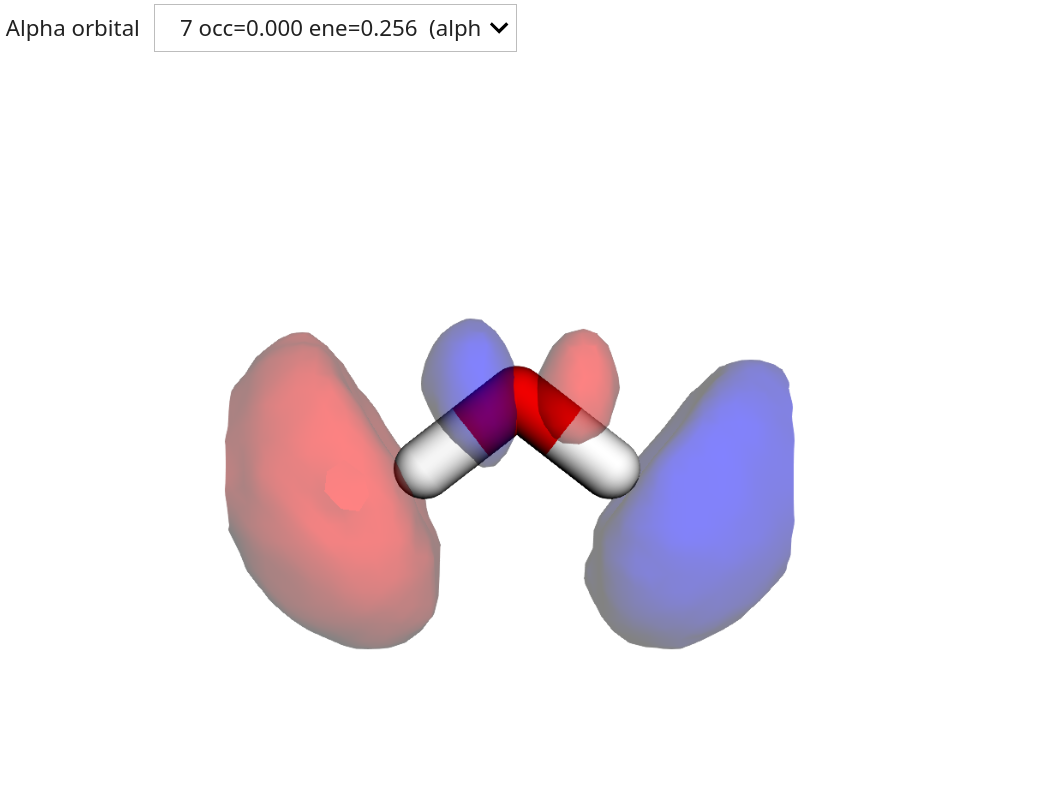
Visualizing molecular orbitals¶
The resulting molecular orbitals (MOs) can be visualized using the OrbitalViewer class.
viewer = vlx.OrbitalViewer()
viewer.plot(molecule, basis, scf_drv.mol_orbs)