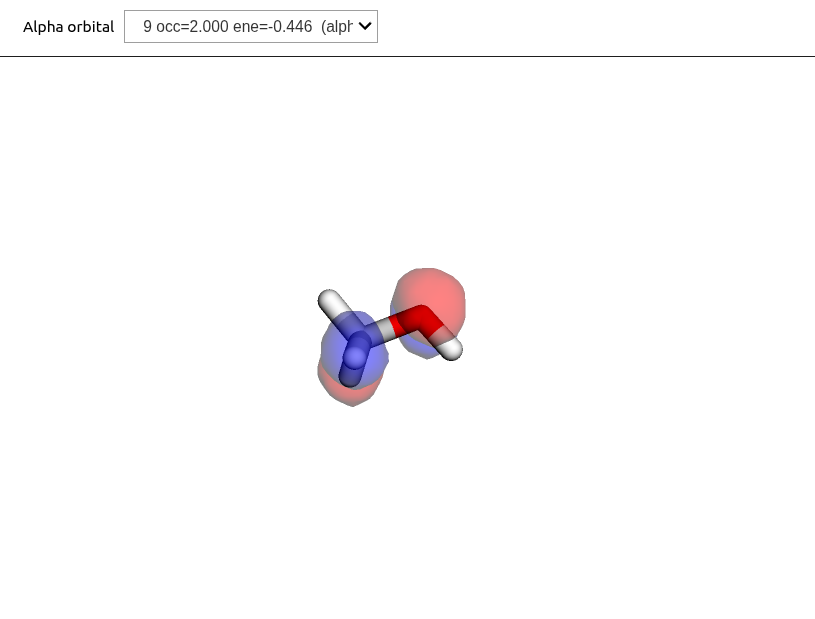
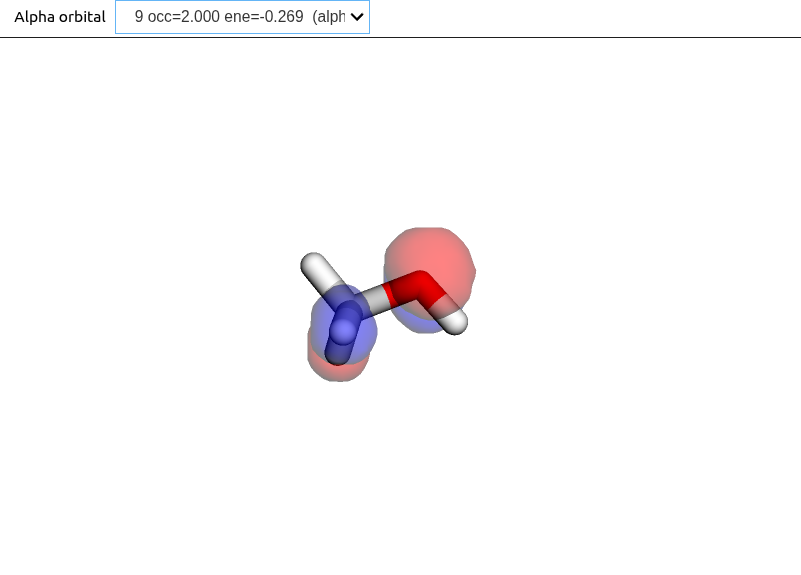
Here we consider the optimization of the electronic structure using Hartree--Fock (HF) and Kohn-Sham density functional theory (KS-DFT), and how to plot the resulting molecular orbitals. Our example is methanol, using the xTB structure from the previous section.
At the HF/6-311G level of theory, the electronic structure is optimized by creating the molecule and basis objects, setting up an unrestricted SCF driver, and performing the SCF calculation:
Self Consistent Field Driver Setup
====================================
Wave Function Model : Spin-Restricted Kohn-Sham
Initial Guess Model : Superposition of Atomic Densities
Convergence Accelerator : Two Level Direct Inversion of Iterative Subspace
Max. Number of Iterations : 50
Max. Number of Error Vectors : 10
Convergence Threshold : 1.0e-06
ERI Screening Threshold : 1.0e-12
Linear Dependence Threshold : 1.0e-06
Exchange-Correlation Functional : B3LYP
Molecular Grid Level : 4
* Info * Using the B3LYP functional.
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch., J. Phys. Chem. 98, 11623 (1994)
* Info * Using the Libxc library (v7.0.0).
S. Lehtola, C. Steigemann, M. J.T. Oliveira, and M. A.L. Marques., SoftwareX 7, 1–5 (2018)
* Info * Using the following algorithm for XC numerical integration.
J. Kussmann, H. Laqua and C. Ochsenfeld, J. Chem. Theory Comput. 2021, 17, 1512-1521
* Info * Starting Reduced Basis SCF calculation...
* Info * ...done. SCF energy in reduced basis set: -115.018935737583 a.u. Time: 0.07 sec.
Iter. | Kohn-Sham Energy | Energy Change | Gradient Norm | Max. Gradient | Density Change
--------------------------------------------------------------------------------------------
1 -115.712788803550 0.0000000000 0.12055772 0.01852446 0.00000000
2 -115.712969331741 -0.0001805282 0.10625363 0.01806455 0.05103107
3 -115.714365718827 -0.0013963871 0.00476261 0.00081966 0.02632569
4 -115.714368563845 -0.0000028450 0.00049882 0.00007594 0.00147822
5 -115.714368569914 -0.0000000061 0.00046205 0.00007408 0.00021746
6 -115.714368587631 -0.0000000177 0.00003830 0.00000547 0.00009628
7 -115.714368587831 -0.0000000002 0.00000282 0.00000036 0.00001735
8 -115.714368587833 -0.0000000000 0.00000034 0.00000006 0.00000156
*** SCF converged in 8 iterations. Time: 0.20 sec.
Spin-Restricted Kohn-Sham:
--------------------------
Total Energy : -115.7143685878 a.u.
Electronic Energy : -156.2327230764 a.u.
Nuclear Repulsion Energy : 40.5183544886 a.u.
------------------------------------
Gradient Norm : 0.0000003430 a.u.
Ground State Information
------------------------
Charge of Molecule : 0.0
Multiplicity (2S+1) : 1
Magnetic Quantum Number (M_S) : 0.0
print(f'The total (B3LYP) energy is {scf_b3lyp.get_scf_energy(): .5f} Hartree')