Core-valence separation#
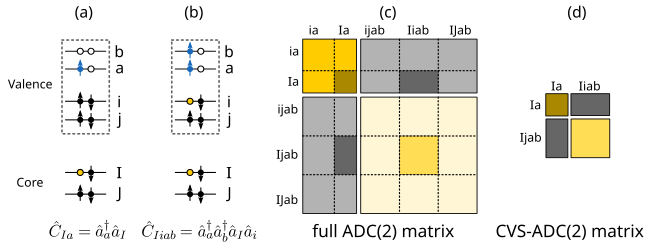
One way to reach the space of core-excitations (and thus be able to compute, for example, X-ray absorption spectra), without having to deal with an intractably large ADC matrix is provided by the core-valence separation (CVS) approximation. CVS is obtained by decoupling the core and valence excitation spaces and is motivated by the large energy separation between them [CDS80]. Essentially, it consists of applying only excitation operators that involve one core electron – see (a) and (b) in below figure. This translates into keeping only those blocks of the ADC matrix which include one core orbital (c) and significantly reduces the size of the matrix to be diagonalized (d). The error introduced by this approximation is small and system independent [HF20].
Illustration of the (a) single excitations and (b) double excitations involved in the CVS approximation. (c) Schematic representation of the full ADC(2) matrix and (d) the size reduction achieved by the CVS approximation.
The CVS approximation allows the computation of X-ray spectroscopies, which are important techniques for the characterisation of materials, providing element-specific probes sensitive to the local environment. In the remaining of this section we will introduce X-ray absorption spectroscopy (XAS) and review some of its features. For a more in-depth discussion see Refs. [Stohr92] and [ND18].
The absorption of soft X-ray electromagnetic radiation by a molecular system results in electronic excitation between core initial electronic states (localized and atomic-like) and final states that are more delocalized, and sometimes continuum-like. A schematic and simplified illustration of this process, alongside the corresponding XAS spectrum is shown below. Here (a) shows a schematic potential and (b) schematic representation of the C K-edge X-ray absorption spectrum for the dichloroethylene molecule (adapted from Ref. [Stohr92], Fig. 4.2.).
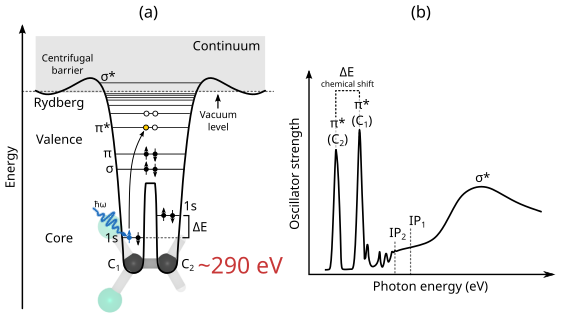
The core-level binding energies of 1s electrons are element-specific, with very large energy separations between different elements, e.g. \(\sim\)290 eV (C), \(\sim\)400 eV (N), and \(\sim\)500 eV (O). Additionally, atoms of the same species placed in different chemical environments have binding energies which differ by a few electron-volts. This means that absorption peaks corresponding to transitions from chemically inequivalent atoms will occur at different photon energies. The separation between these peaks is called chemical shift and it enables the use of XAS for chemical analysis and materials characterization.
In the case of core-excitations, electron–electron correlation and the presence of the core-hole influence the excitation energies and transition probabilities. These relaxation effects are present also in the case of valence-excitations, but they are more pronounced in the case of core-excitations. This is due the presence of a localized core-hole, which leads to a strong net attraction of the electron density towards the probed atom. In addition, the interaction with the excited electron creates a small repulsive polarisation effect in the valence region. Both these two counteracting effects need to be accounted for in order to accurately model XAS. At the ADC level of theory, this is achieved by including at least doubly excited configurations in the construction of the ADC matrix.