Cheat sheet#
Here we provide quick reference calculation for some of the most common X-ray spectrum calculations, considering XPS/IE, XAS, and (non-resonant) XES of water, as well as a typical workflow.
Typical workflow#
Perform structure optimization
Calculate the spectra/ionization energies
Choose suitable level of theory
Depending on spectroscopy:
Ionization energy: using \(\Delta\)-methods, or target transitions to extremely diffuse MOs
XES: ADC, TDSCF, and using ground state MOs
Analysis and assignment of the spectra
Looking at amplitudes
Visualization
IEs and XPS#
Koopmans’ theorem#
While it is not recommended for any production calculations, estimates of ionization energies can be obtained from Koopmans’ theorem:
import numpy as np
import veloxchem as vlx
# for vlx
silent_ostream = vlx.OutputStream(None)
from mpi4py import MPI
comm = MPI.COMM_WORLD
# au to eV conversion factor
au2ev = 27.211386
water_mol_str = """
O 0.0000000000 0.0000000000 0.1178336003
H -0.7595754146 -0.0000000000 -0.4713344012
H 0.7595754146 0.0000000000 -0.4713344012
"""
# Create veloxchem mol and basis objects
mol_vlx = vlx.Molecule.read_molecule_string(water_mol_str)
bas_vlx = vlx.MolecularBasis.read(mol_vlx, "6-31G")
# Perform SCF calculation
scf_gs = vlx.ScfRestrictedDriver(comm, ostream=silent_ostream)
scf_results = scf_gs.compute(mol_vlx, bas_vlx)
# Extract orbital energies
orbital_energies = scf_results["E_alpha"]
print("1s E from Koopmans' theorem:", np.around(au2ev * orbital_energies[0], 2), "eV")
1s E from Koopmans' theorem: -559.5 eV
\(\Delta\)-methods#
Substantially improved ionization energies are obtained using \(\Delta\)-methods, where the energy difference of the ground state and core-hole state is used to estimate the IE:
Note
pyscf version - to be changed
import copy
import numpy as np
from pyscf import gto, mp, scf
water_xyz = """
O 0.0000000000 0.0000000000 0.1178336003
H -0.7595754146 -0.0000000000 -0.4713344012
H 0.7595754146 0.0000000000 -0.4713344012
"""
# Create pyscf mol object
mol = gto.Mole()
mol.atom = water_xyz
mol.basis = "6-31G"
mol.build()
# Perform unrestricted SCF calculation
scf_gs = scf.UHF(mol)
scf_gs.kernel()
# Copy molecular orbitals and occupations
mo0 = copy.deepcopy(scf_gs.mo_coeff)
occ0 = copy.deepcopy(scf_gs.mo_occ)
# Create 1s core-hole by setting alpha_0 population to zero
occ0[0][0] = 0.0
# Perform unrestricted SCF calculation with MOM constraint
scf_ion = scf.UHF(mol)
scf.addons.mom_occ(scf_ion, mo0, occ0)
scf_ion.kernel()
# Run MP2 on neutral and core-hole state
mp_res = mp.MP2(scf_gs).run()
mp_ion = mp.MP2(scf_ion).run()
# IE from energy difference
print(
"HF ionization energy:",
np.around(au2ev * (scf_ion.energy_tot() - scf_gs.energy_tot()), 2),
"eV",
)
print(
"MP2 ionzation energy:", np.around(au2ev * (mp_ion.e_tot - mp_res.e_tot), 2), "eV"
)
XAS#
CVS-ADC#
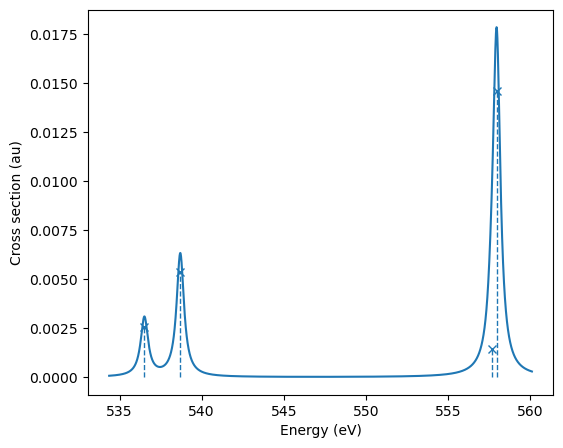
Absorption spectra can be calculated using CVS-ADC:
import gator
import matplotlib.pyplot as plt
water_mol_str = """
O 0.0000000000 0.0000000000 0.1178336003
H -0.7595754146 -0.0000000000 -0.4713344012
H 0.7595754146 0.0000000000 -0.4713344012
"""
# Construct structure and basis objects
struct = gator.get_molecule(water_mol_str)
basis = gator.get_molecular_basis(struct, "6-31G")
# Perform SCF calculation
scf_gs = gator.run_scf(struct, basis)
# Calculate the 6 lowest eigenstates with CVS restriction to MO #1 (oxygen 1s)
adc_res = gator.run_adc(
struct, basis, scf_gs, method="cvs-adc2x", singlets=4, core_orbitals=1
)
# Print information on eigenstates
print(adc_res.describe())
plt.figure(figsize=(6, 5))
# Convolute using functionalities available in gator and adcc
adc_res.plot_spectrum()
plt.show()
+--------------------------------------------------------------+
| cvs-adc2x singlet , converged |
+--------------------------------------------------------------+
| # excitation energy osc str |v1|^2 |v2|^2 |
| (au) (eV) |
| 0 19.71638 536.5099 0.0178 0.8 0.2 |
| 1 19.7967 538.6956 0.0373 0.8087 0.1913 |
| 2 20.49351 557.6567 0.0099 0.7858 0.2142 |
| 3 20.50482 557.9647 0.1016 0.8441 0.1559 |
+--------------------------------------------------------------+
CPP-DFT#
To be added
XES#
ADC#
The non-resonant X-ray emission spectrum can be calculated with a two-step approach using ADC:
Note
pyscf version - to be changed
import copy
import adcc
import matplotlib.pyplot as plt
import numpy as np
from pyscf import gto, mp, scf
water_xyz = """
O 0.0000000000 0.0000000000 0.1178336003
H -0.7595754146 -0.0000000000 -0.4713344012
H 0.7595754146 0.0000000000 -0.4713344012
"""
# Create pyscf mol object
mol = gto.Mole()
mol.atom = water_xyz
mol.basis = "6-31G"
mol.build()
# Perform unrestricted SCF calculation
scf_res = scf.UHF(mol)
scf_res.kernel()
# Copy molecular orbitals
mo0 = copy.deepcopy(scf_res.mo_coeff)
occ0 = copy.deepcopy(scf_res.mo_occ)
# Create 1s core-hole by setting alpha_0 population to zero
occ0[0][0] = 0.0
# Perform unrestricted SCF calculation with MOM constraint
scf_ion = scf.UHF(mol)
scf.addons.mom_occ(scf_ion, mo0, occ0)
scf_ion.kernel()
# Perform ADC calculation
adc_xes = adcc.adc2(scf_ion, n_states=4)
# Print information on eigenstates
print(adc_xes.describe())
plt.figure(figsize=(6, 5))
# Convolute using functionalities available in gator and adcc
adc_xes.plot_spectrum()
plt.show()
Ground state MOs#
To be added, using the approach here.