State-averaged MCSCF#
CASCI response#
As for truncated CI, the exact state response formulas also apply to CASCI and other active space CIs. The method can thus create all excited states that arise from excitations within the active space. This can also be used as an approximation for a MCSCF response, neglecting the orbital part of the response.
Let us try it on our usual water case. We’ll use an active space including the 2 pairs of O-H \(\sigma\) and \(\sigma^*\) (4 electrons in 4 orbitals). However, since the main excitation also involves the oxygen lone pair, we add this orbital too, for a total CAS(6,5).
First, we try the intuitive concept of first optimizing the CASSCF wavefunction for the ground state, and then applying CASCI response with the 5 states.
import veloxchem as vlx
import multipsi as mtp
import numpy as np
mol_str = """3
O 0.0000000000 0.0000000000 0.1178336003
H -0.7595754146 -0.0000000000 -0.4713344012
H 0.7595754146 0.0000000000 -0.4713344012
"""
molecule = vlx.Molecule.read_xyz_string(mol_str)
basis = vlx.MolecularBasis.read(molecule, "6-31g")
scf_drv = vlx.ScfRestrictedDriver()
scf_results = scf_drv.compute(molecule, basis)
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
# CAS(6,5) calculation of water
space=mtp.OrbSpace(molecule,scf_drv.mol_orbs)
space.cas(6,5) #3 O_2p and 2 H_1s
# CASSCF calculation
mcscf_drv=mtp.McscfDriver()
mcscf_results = mcscf_drv.compute(molecule,basis,space)
nstates=5
# CASCI excitation energies
cidrv=mtp.CIDriver()
ci_results = cidrv.compute(molecule,basis,space,nstates)
# Transition properties
SI = mtp.StateInteraction()
si_results = SI.compute(molecule, basis, ci_results)
# Results computed in CI response
FCI_excitation_energies=[8.44992, 10.68451, 10.97654, 13.37692]
CIS_excitation_energies=[9.38059, 11.30739, 11.83172, 13.90237]
au2ev = 27.211386
print("Full CI Energies")
print(FCI_excitation_energies)
print("CIS Energies")
print(CIS_excitation_energies)
print("CASCI Energies")
print(au2ev*np.array(si_results['energies']))
Full CI Energies
[8.44992, 10.68451, 10.97654, 13.37692]
CIS Energies
[9.38059, 11.30739, 11.83172, 13.90237]
CASCI Energies
[18.62048309 18.73205969 26.61689981 28.51722467]
As we can see, the result is very disappointing, very far from the full CI result, and even from the CIS result. The reason for that is the orbitals. The orbitals were optimized for the ground state, and not for the excited states. Like for truncated CI, we thus have an imbalance between the description of the ground and excited states leading to (significantly) too high excitation energies. In particular, in this case, the \(\sigma^*\) orbitals are optimal to describe correlation in the O-H bonds, but not for the electronic excitation.
State-specific optimization#
A way to improve over the previous situation is to optimize every state separately with their own orbitals. This is called state-specific optimization. To ensure that we are doing an excited state, we still need to compute the ground states (or in general all states below the targeted state), but tell the program to optimize the orbitals specifically for the n_th state. In MultiPsi, this is done by providing “weights” (this concept will become more clear in the next section), a weight of 1 for the state of interest and 0 for all the other, meaning they will not influence the orbital optimization.
mcscf_results = mcscf_drv.compute(molecule,basis,space)
e0 = mcscf_results["energies"][0]
mcscf_results = mcscf_drv.compute(molecule,basis,space.copy(), [0,1])
e1 = mcscf_results["energies"][1]
Multi-Configurational Self-Consistent Field Driver
====================================================
Active space definition:
------------------------
Number of inactive (occupied) orbitals: 2
Number of active orbitals: 5
Number of virtual orbitals: 6
This is a CASSCF wavefunction: CAS(6,5)
CI expansion:
-------------
Number of determinants: 100
╭────────────────────────────────────╮
│ Driver settings │
╰────────────────────────────────────╯
Number of states : 1
State-specific calculation
- State of interest : 1
Max. iterations : 50
BFGS window : 5
Convergence thresholds:
- Energy change : 1e-08
- Gradient sq. norm : 1e-08
MCSCF Iterations
-------------------
Iter. | Average Energy | E. Change | Grad. Norm | CI Iter. | Trust rad. | Time
---------------------------------------------------------------------------------
1 -76.037843341 0.0e+00 7.9e-11 0 0.40 0:00:00
** Convergence reached in 1 iterations
2 -76.037843341 -4.8e-11 7.3e-12 0 0.40 0:00:00
Final results
-------------
* State 1
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -76.0378433409989
- Natural orbitals
1.99931 1.97806 1.97495 0.02400 0.02368
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 1:
--------------------------
Occupation: 2.000 Energy: -20.55484 a.u.
( 1 O 1s : -1.00)
Molecular Orbital No. 2:
--------------------------
Occupation: 2.000 Energy: -1.07302 a.u.
( 1 O 1s : 0.22) ( 1 O 2s : -0.48) ( 1 O 3s : -0.57)
( 1 O 1p0 : -0.24) ( 1 O 2p0 : -0.19)
Molecular Orbital No. 3:
--------------------------
Occupation: 1.999 Energy: -0.50216 a.u.
( 1 O 1p-1: -0.64) ( 1 O 2p-1: -0.51)
Molecular Orbital No. 4:
--------------------------
Occupation: 1.978 Energy: -0.83590 a.u.
( 1 O 1p0 : -0.51) ( 1 O 2p0 : -0.35) ( 2 H 1s : 0.21)
( 3 H 1s : 0.21)
Molecular Orbital No. 5:
--------------------------
Occupation: 1.975 Energy: -0.70220 a.u.
( 1 O 1p+1: 0.51) ( 1 O 2p+1: 0.25) ( 2 H 1s : -0.27)
( 3 H 1s : 0.27)
Molecular Orbital No. 6:
--------------------------
Occupation: 0.024 Energy: 0.75291 a.u.
( 1 O 1p+1: -0.87) ( 2 H 1s : -0.46) ( 2 H 2s : -0.30)
( 3 H 1s : 0.46) ( 3 H 2s : 0.30)
Molecular Orbital No. 7:
--------------------------
Occupation: 0.024 Energy: 0.73045 a.u.
( 1 O 2s : -0.50) ( 1 O 3s : -0.17) ( 1 O 1p0 : 0.77)
( 1 O 2p0 : -0.18) ( 2 H 1s : 0.43) ( 2 H 2s : 0.20)
( 3 H 1s : 0.43) ( 3 H 2s : 0.20)
Molecular Orbital No. 8:
--------------------------
Occupation: 0.000 Energy: 0.68035 a.u.
( 1 O 2s : 0.19) ( 1 O 3s : -1.21) ( 1 O 1p0 : -0.41)
( 1 O 2p0 : 0.76) ( 2 H 1s : -0.40) ( 2 H 2s : 1.13)
( 3 H 1s : -0.40) ( 3 H 2s : 1.13)
Molecular Orbital No. 9:
--------------------------
Occupation: 0.000 Energy: 0.78475 a.u.
( 1 O 1p+1: 0.28) ( 1 O 2p+1: -0.73) ( 2 H 1s : 0.62)
( 2 H 2s : -1.60) ( 3 H 1s : -0.62) ( 3 H 2s : 1.60)
Dipole moment for state 1
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.981838 a.u. -2.495584 Debye
Total : 0.981838 a.u. 2.495584 Debye
Total MCSCF time: 00:00:00
Multi-Configurational Self-Consistent Field Driver
====================================================
Active space definition:
------------------------
Number of inactive (occupied) orbitals: 2
Number of active orbitals: 5
Number of virtual orbitals: 6
This is a CASSCF wavefunction: CAS(6,5)
CI expansion:
-------------
Number of determinants: 100
╭────────────────────────────────────╮
│ Driver settings │
╰────────────────────────────────────╯
Number of states : 2
State-specific calculation
- State of interest : 2
Max. iterations : 50
BFGS window : 5
Convergence thresholds:
- Energy change : 1e-08
- Gradient sq. norm : 1e-08
MCSCF Iterations
-------------------
Iter. | Average Energy | E. Change | Grad. Norm | CI Iter. | Trust rad. | Time
---------------------------------------------------------------------------------
1 -75.353554617 0.0e+00 1.6e-01 0 0.40 0:00:00
2 -75.593568983 -2.4e-01 4.2e-02 0 0.40 0:00:00
3 -75.632460021 -3.9e-02 1.1e-02 0 0.56 0:00:00
4 -75.642345164 -9.9e-03 1.1e-04 0 0.56 0:00:00
5 -75.642439232 -9.4e-05 1.4e-05 0 0.56 0:00:00
6 -75.642461616 -2.2e-05 2.1e-06 0 0.67 0:00:00
7 -75.642463966 -2.3e-06 5.8e-07 0 0.80 0:00:00
8 -75.642464740 -7.7e-07 8.6e-08 0 0.80 0:00:00
9 -75.642464800 -6.0e-08 1.7e-08 0 0.80 0:00:00
10 -75.642464810 -1.1e-08 1.1e-09 0 0.80 0:00:00
11 -75.642464811 -9.0e-10 1.7e-10 0 0.80 0:00:00
** Convergence reached in 11 iterations
12 -75.642464811 -1.2e-10 2.0e-11 0 0.80 0:00:00
Final results
-------------
* State 1
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.99867538837437
- Natural orbitals
1.98064 1.98062 0.01721 1.99899 0.02255
* State 2
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -75.64246481146712
- Natural orbitals
1.99396 1.97991 1.00091 1.00000 0.02521
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 1:
--------------------------
Occupation: 2.000 Energy: -20.71709 a.u.
( 1 O 1s : 1.00)
Molecular Orbital No. 2:
--------------------------
Occupation: 2.000 Energy: -1.18833 a.u.
( 1 O 1s : -0.21) ( 1 O 2s : 0.50) ( 1 O 3s : 0.54)
( 1 O 1p0 : 0.27) ( 1 O 2p0 : 0.19)
Molecular Orbital No. 3:
--------------------------
Occupation: 1.994 Energy: -0.84981 a.u.
( 1 O 1p+1: 0.59) ( 1 O 2p+1: 0.31) ( 2 H 1s : -0.22)
( 3 H 1s : 0.22)
Molecular Orbital No. 4:
--------------------------
Occupation: 1.980 Energy: -0.98576 a.u.
( 1 O 2s : 0.17) ( 1 O 1p0 : -0.54) ( 1 O 2p0 : -0.34)
( 2 H 1s : 0.18) ( 3 H 1s : 0.18)
Molecular Orbital No. 5:
--------------------------
Occupation: 1.001 Energy: 0.12396 a.u.
( 1 O 1p+1: 0.37) ( 1 O 2p+1: 0.71) ( 2 H 1s : 0.16)
( 2 H 2s : 1.25) ( 3 H 1s : -0.16) ( 3 H 2s : -1.25)
Molecular Orbital No. 6:
--------------------------
Occupation: 1.000 Energy: -0.35135 a.u.
( 1 O 1p-1: -0.70) ( 1 O 2p-1: -0.44)
Molecular Orbital No. 7:
--------------------------
Occupation: 0.025 Energy: 0.52545 a.u.
( 1 O 2s : 0.42) ( 1 O 3s : 0.41) ( 1 O 1p0 : -0.67)
( 2 H 1s : -0.47) ( 2 H 2s : -0.33) ( 3 H 1s : -0.47)
( 3 H 2s : -0.33)
Molecular Orbital No. 8:
--------------------------
Occupation: 0.000 Energy: 0.73663 a.u.
( 1 O 3s : -1.03) ( 1 O 1p0 : -0.49) ( 1 O 2p0 : 0.75)
( 2 H 1s : -0.45) ( 2 H 2s : 1.06) ( 3 H 1s : -0.45)
( 3 H 2s : 1.06)
Molecular Orbital No. 9:
--------------------------
Occupation: 0.000 Energy: 0.99710 a.u.
( 1 O 2p+1: 0.65) ( 2 H 1s : 0.97) ( 2 H 2s : -0.62)
( 3 H 1s : -0.97) ( 3 H 2s : 0.62)
Dipole moment for state 1
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : 0.000000 a.u. 0.000000 Debye
Z : -0.878313 a.u. -2.232449 Debye
Total : 0.878313 a.u. 2.232449 Debye
Dipole moment for state 2
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.001754 a.u. -0.004459 Debye
Total : 0.001754 a.u. 0.004459 Debye
Total MCSCF time: 00:00:01
print(f"First state-specific MCSCF excitation energy = {(e1-e0)*au2ev:.4f}")
print(f"Corresponding (2nd) FCI excitation energy = {FCI_excitation_energies[1]:.4f}")
First state-specific MCSCF excitation energy = 10.7588
Corresponding (2nd) FCI excitation energy = 10.6845
As hoped, the first excitation energy is now a much better match to the FCI result. Actually, if we analyzed the orbitals, we would see that the state we converged is actually the second excited state, and the result is thus very close to the FCI result. We can also note that the second calculation still prints the ground state energy, but on the orbitals optimized for the excited state, and because of this, the energy of this state is higher than in the first calculation.
While in principle the most accurate description of the state that can be obtained at the MCSCF level, state-specific optimization has severe shortcomings hindering broader applicability. First, they way the calculation is done, the excited state is not necessarily orthogonal to the ground state, and it may therefore be artificially lower than it should. Second, if there is a small energy gap between states, the optimization may have trouble converging. We can see it if we try to optimize a higher excited state:
mcscf_results = mcscf_drv.compute(molecule,basis,space.copy(), [0,0,0,1])
Multi-Configurational Self-Consistent Field Driver
====================================================
Active space definition:
------------------------
Number of inactive (occupied) orbitals: 2
Number of active orbitals: 5
Number of virtual orbitals: 6
This is a CASSCF wavefunction: CAS(6,5)
CI expansion:
-------------
Number of determinants: 100
╭────────────────────────────────────╮
│ Driver settings │
╰────────────────────────────────────╯
Number of states : 4
State-specific calculation
- State of interest : 4
Max. iterations : 50
BFGS window : 5
Convergence thresholds:
- Energy change : 1e-08
- Gradient sq. norm : 1e-08
MCSCF Iterations
-------------------
Iter. | Average Energy | E. Change | Grad. Norm | CI Iter. | Trust rad. | Time
---------------------------------------------------------------------------------
1 -75.059689492 0.0e+00 1.8e-01 0 0.40 0:00:00
2 -75.289477082 -2.3e-01 1.9e-01 0 0.40 0:00:00
3 -75.512738636 -2.2e-01 1.2e-01 0 0.56 0:00:00
4 -75.540095358 -2.7e-02 9.0e-02 0 0.56 0:00:00
5 -75.590325896 -5.0e-02 2.9e-02 0 0.39 0:00:00
6 -75.600770537 -1.0e-02 7.5e-02 0 0.39 0:00:00
7 -75.603745659 -3.0e-03 5.8e-03 0 0.39 0:00:00
8 -75.606581228 -2.8e-03 2.5e-03 0 0.27 0:00:00
9 -75.603872438 2.7e-03 3.3e-02 0 0.38 0:00:00
10 -75.609337925 -5.5e-03 2.2e-03 0 0.19 0:00:00
11 -75.586334602 2.3e-02 4.4e-02 0 0.19 0:00:00
12 -75.608208824 -2.2e-02 2.3e-02 0 0.10 0:00:00
13 -75.595640388 1.3e-02 2.7e-02 0 0.13 0:00:00
14 -75.606460065 -1.1e-02 4.8e-03 0 0.07 0:00:00
15 -75.610468047 -4.0e-03 1.4e-03 0 0.08 0:00:00
16 -75.572806443 3.8e-02 4.9e-01 0 0.10 0:00:00
17 -75.596176427 -2.3e-02 2.3e-01 0 0.05 0:00:00
18 -75.593186060 3.0e-03 3.3e-01 0 0.07 0:00:00
19 -75.608532025 -1.5e-02 1.3e-02 0 0.04 0:00:00
20 -75.610679742 -2.1e-03 9.4e-03 0 0.05 0:00:00
21 -75.592808452 1.8e-02 1.0e-01 0 0.07 0:00:00
22 -75.605169953 -1.2e-02 3.7e-02 0 0.04 0:00:00
23 -75.609771321 -4.6e-03 3.8e-03 0 0.06 0:00:00
24 -75.610649741 -8.8e-04 1.8e-03 0 0.06 0:00:00
25 -75.605341026 5.3e-03 8.0e-02 0 0.06 0:00:00
26 -75.611292802 -6.0e-03 1.2e-03 0 0.04 0:00:00
27 -75.610598846 6.9e-04 9.1e-03 0 0.04 0:00:00
28 -75.611430414 -8.3e-04 8.2e-04 0 0.04 0:00:00
29 -75.604576504 6.9e-03 6.9e-02 0 0.05 0:00:00
30 -75.611151268 -6.6e-03 1.6e-02 0 0.04 0:00:00
31 -75.608798835 2.4e-03 6.2e-03 0 0.05 0:00:00
32 -75.610070132 -1.3e-03 1.9e-03 0 0.04 0:00:00
33 -75.610931707 -8.6e-04 3.5e-03 0 0.04 0:00:00
34 -75.602994069 7.9e-03 8.7e-02 0 0.04 0:00:00
35 -75.611206788 -8.2e-03 1.2e-03 0 0.04 0:00:00
36 -75.610538616 6.7e-04 2.8e-03 0 0.05 0:00:00
37 -75.611104135 -5.7e-04 1.1e-03 0 0.04 0:00:00
38 -75.607135046 4.0e-03 4.2e-02 0 0.04 0:00:00
39 -75.611289988 -4.2e-03 8.4e-04 0 0.04 0:00:00
40 -75.611358595 -6.9e-05 8.0e-04 0 0.04 0:00:00
41 -75.604465711 6.9e-03 9.5e-02 0 0.04 0:00:00
42 -75.611461803 -7.0e-03 6.6e-03 0 0.04 0:00:00
43 -75.610546749 9.2e-04 1.2e-03 0 0.05 0:00:00
44 -75.610281550 2.7e-04 3.8e-03 0 0.04 0:00:00
45 -75.611171592 -8.9e-04 2.0e-03 0 0.04 0:00:00
46 -75.601715612 9.5e-03 7.2e-02 0 0.05 0:00:00
47 -75.610573728 -8.9e-03 2.5e-02 0 0.04 0:00:00
48 -75.598748591 1.2e-02 2.2e-01 0 0.05 0:00:00
49 -75.605339430 -6.6e-03 7.8e-02 0 0.04 0:00:00
50 -75.608736976 -3.4e-03 1.9e-02 0 0.05 0:00:00
** Convergence not reached after 50 iterations
Final results
-------------
* State 1
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.96501757187585
- Natural orbitals
1.99957 1.98440 0.00676 1.99314 0.01612
* State 2
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.70270319216573
- Natural orbitals
1.00048 1.98753 1.99732 0.99845 0.01622
* State 3
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.61741109477028
- Natural orbitals
1.01385 1.99526 1.98208 0.02478 0.98404
* State 4
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -75.60873697571489
- Natural orbitals
1.96048 1.99564 1.20245 0.79517 0.04625
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 1:
--------------------------
Occupation: 2.000 Energy: -20.71949 a.u.
( 1 O 1s : -1.00)
Molecular Orbital No. 2:
--------------------------
Occupation: 2.000 Energy: -1.45314 a.u.
( 1 O 1s : 0.21) ( 1 O 2s : -0.49) ( 1 O 3s : -0.47)
( 1 O 1p0 : 0.16)
Molecular Orbital No. 3:
--------------------------
Occupation: 1.996 Energy: -0.69310 a.u.
( 1 O 1p+1: 0.29) ( 1 O 1p-1: 0.59) ( 1 O 2p+1: 0.15)
( 1 O 2p-1: 0.40)
Molecular Orbital No. 4:
--------------------------
Occupation: 1.960 Energy: -0.76984 a.u.
( 1 O 1p+1: -0.48) ( 1 O 1p-1: 0.34) ( 1 O 2p+1: -0.25)
( 1 O 2p-1: 0.24) ( 2 H 1s : 0.20) ( 3 H 1s : -0.20)
Molecular Orbital No. 5:
--------------------------
Occupation: 1.202 Energy: -0.29127 a.u.
( 1 O 3s : 0.48) ( 1 O 1p0 : -0.60) ( 1 O 2p0 : -0.58)
( 2 H 2s : -0.55) ( 3 H 2s : -0.56)
Molecular Orbital No. 6:
--------------------------
Occupation: 0.795 Energy: -0.13448 a.u.
( 1 O 2s : 0.28) ( 1 O 3s : 1.00) ( 1 O 1p0 : 0.23)
( 2 H 1s : -0.16) ( 2 H 2s : -0.76) ( 3 H 1s : -0.15)
( 3 H 2s : -0.78)
Molecular Orbital No. 7:
--------------------------
Occupation: 0.046 Energy: 0.25788 a.u.
( 1 O 1p+1: 0.45) ( 1 O 2p+1: 0.67) ( 2 H 1s : 0.18)
( 2 H 2s : 1.20) ( 3 H 1s : -0.18) ( 3 H 2s : -1.18)
Molecular Orbital No. 8:
--------------------------
Occupation: 0.000 Energy: 0.99214 a.u.
( 1 O 2p+1: 0.70) ( 2 H 1s : 0.97) ( 2 H 2s : -0.61)
( 3 H 1s : -0.96) ( 3 H 2s : 0.60)
Molecular Orbital No. 9:
--------------------------
Occupation: 0.000 Energy: 1.04190 a.u.
( 1 O 1p-1: 0.94) ( 1 O 2p-1: -1.06)
Dipole moment for state 1
---------------------------
X : -0.080523 a.u. -0.204670 Debye
Y : -0.002618 a.u. -0.006655 Debye
Z : 2.039047 a.u. 5.182741 Debye
Total : 2.040638 a.u. 5.186785 Debye
Dipole moment for state 2
---------------------------
X : -0.081469 a.u. -0.207075 Debye
Y : -0.007144 a.u. -0.018158 Debye
Z : 1.451095 a.u. 3.688315 Debye
Total : 1.453398 a.u. 3.694168 Debye
Dipole moment for state 3
---------------------------
X : -0.013867 a.u. -0.035246 Debye
Y : -0.007749 a.u. -0.019696 Debye
Z : 2.734143 a.u. 6.949499 Debye
Total : 2.734190 a.u. 6.949617 Debye
Dipole moment for state 4
---------------------------
X : -0.087225 a.u. -0.221704 Debye
Y : -0.003325 a.u. -0.008451 Debye
Z : 0.166119 a.u. 0.422232 Debye
Total : 0.187656 a.u. 0.476974 Debye
Total MCSCF time: 00:00:06
The optimization oscillates as optimizing the second excited state brings its energy very close to the first, and thus the order of the states keep switching. While there exists “root following” techniques to overcome this, state-specific optimizations are usually fairly cumbersome.
For all these reasons, state-specific optimization is usually replaced by a so-called state-averaged optimization.
State-averaging#
Ideally, we would want each state to have its own orbitals while maintaining orthogonality betweem states. In the next chapter, we will see how this can be achieved within the framework of full MCSCF response. However, there is a way to improve the result without including the orbital response. The concept is to use only one orbital set for all states, but to make these orbitals a “compromise” between the needs of the ground state and that of the excited states. The description of each state would be worse but the hope is that this error would be consistent across all states and mostly cancel out in the excitation energies.
The way to do this is to construct a “state-average” MCSCF (SA-MCSCF), that is optimize the orbitals to minimize the average energy of all states. Doing this requires only a trivial modification of the code, namely the CI part of the MCSCF includes all states (here 5) and then the MCSCF energy is replaced by the average energy of all states and the CI densities are replaced by the average density of all states. Note that this is simply an extension of the state-specific optimization mentioned above, but using equal weights for all states.
Let’s see it in practice:
# CASSCF calculation
# 5 states included already in the MCSCF.
mcscf_results = mcscf_drv.compute(molecule,basis,space,nstates)
# Transition properties
si_results=SI.compute(molecule, basis, mcscf_results)
print("Full CI Energies")
print(FCI_excitation_energies)
print("CIS Energies")
print(CIS_excitation_energies)
print("SA-CASSCF Energies")
print(au2ev*np.array(si_results['energies']))
Full CI Energies
[8.44992, 10.68451, 10.97654, 13.37692]
CIS Energies
[9.38059, 11.30739, 11.83172, 13.90237]
SA-CASSCF Energies
[ 7.55787633 9.83565757 10.32867791 12.5632305 ]
The result is now significantly closer to the converged full CI result. In this particular case, the result is not better than CIS, but of course, one the main motivation to use MCSCF is for systems with strong correlation, which was not the case here.
This method is of course not without issues. In particular, unlike standard responses calculation, the introduction of state-averaging means that the excitation energies depend on the total number of states, since the state-averaged orbitals depend on all states. In particular, we can expect the results to deteriorate with increasing number of states as the orbitals begin to compromise many states of widely different natures:
first_exc_energy=[]
for nstates in range(2,6):
# CASSCF calculation
# 5 states included already in the MCSCF
mcscf_results = mcscf_drv.compute(molecule,basis,space,nstates)
# Transition properties
si_results=SI.compute(molecule, basis, mcscf_results)
first_exc_energy.append(au2ev*si_results['energies'][0])
Show code cell output
Multi-Configurational Self-Consistent Field Driver
====================================================
Active space definition:
------------------------
Number of inactive (occupied) orbitals: 2
Number of active orbitals: 5
Number of virtual orbitals: 6
This is a CASSCF wavefunction: CAS(6,5)
CI expansion:
-------------
Number of determinants: 100
╭────────────────────────────────────╮
│ Driver settings │
╰────────────────────────────────────╯
Number of states : 2
State-averaged calculation
- Equal-weights
Max. iterations : 50
BFGS window : 5
Convergence thresholds:
- Energy change : 1e-08
- Gradient sq. norm : 1e-08
MCSCF Iterations
-------------------
Iter. | Average Energy | E. Change | Grad. Norm | CI Iter. | Trust rad. | Time
---------------------------------------------------------------------------------
1 -75.843270570 0.0e+00 5.4e-03 0 0.40 0:00:00
2 -75.858829181 -1.6e-02 1.9e-03 0 0.40 0:00:00
3 -75.864455912 -5.6e-03 2.8e-04 0 0.48 0:00:00
4 -75.864858792 -4.0e-04 3.6e-05 0 0.48 0:00:00
5 -75.864894173 -3.5e-05 4.6e-06 0 0.58 0:00:00
6 -75.864897104 -2.9e-06 2.4e-07 0 0.69 0:00:00
7 -75.864897238 -1.3e-07 6.9e-09 0 0.80 0:00:00
8 -75.864897243 -4.9e-09 1.8e-10 0 0.80 0:00:00
** Convergence reached in 8 iterations
9 -75.864897243 -2.0e-10 1.2e-11 0 0.80 0:00:00
Final results
-------------
* State 1
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -76.01351106913249
- Natural orbitals
1.98571 1.97737 1.99925 0.01512 0.02254
* State 2
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.71628341744072
- Natural orbitals
1.99623 1.97627 1.00000 1.00146 0.02604
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 1:
--------------------------
Occupation: 2.000 Energy: -20.64857 a.u.
( 1 O 1s : 1.00)
Molecular Orbital No. 2:
--------------------------
Occupation: 2.000 Energy: -1.19269 a.u.
( 1 O 1s : -0.22) ( 1 O 2s : 0.51) ( 1 O 3s : 0.56)
( 1 O 1p0 : 0.22) ( 1 O 2p0 : 0.16)
Molecular Orbital No. 3:
--------------------------
Occupation: 1.985 Energy: -0.86834 a.u.
( 1 O 1p0 : -0.55) ( 1 O 2p0 : -0.35) ( 2 H 1s : 0.19)
( 3 H 1s : 0.19)
Molecular Orbital No. 4:
--------------------------
Occupation: 1.973 Energy: -0.76522 a.u.
( 1 O 1p+1: 0.54) ( 1 O 2p+1: 0.27) ( 2 H 1s : -0.25)
( 3 H 1s : 0.25)
Molecular Orbital No. 5:
--------------------------
Occupation: 1.500 Energy: -0.43591 a.u.
( 1 O 1p-1: 0.66) ( 1 O 2p-1: 0.49)
Molecular Orbital No. 6:
--------------------------
Occupation: 0.515 Energy: 0.12362 a.u.
( 1 O 2s : -0.18) ( 1 O 3s : -1.04) ( 1 O 1p0 : 0.33)
( 1 O 2p0 : 0.38) ( 2 H 2s : 0.89) ( 3 H 2s : 0.89)
Molecular Orbital No. 7:
--------------------------
Occupation: 0.028 Energy: 0.60891 a.u.
( 1 O 1p+1: -0.82) ( 2 H 1s : -0.43) ( 2 H 2s : -0.44)
( 3 H 1s : 0.43) ( 3 H 2s : 0.44)
Molecular Orbital No. 8:
--------------------------
Occupation: 0.000 Energy: 0.84500 a.u.
( 1 O 1p+1: -0.29) ( 1 O 2p+1: 0.56) ( 2 H 1s : -0.70)
( 2 H 2s : 1.51) ( 3 H 1s : 0.70) ( 3 H 2s : -1.51)
Molecular Orbital No. 9:
--------------------------
Occupation: 0.000 Energy: 1.10548 a.u.
( 1 O 1p-1: -0.95) ( 1 O 2p-1: 1.05)
Dipole moment for state 1
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.914415 a.u. -2.324211 Debye
Total : 0.914415 a.u. 2.324211 Debye
Dipole moment for state 2
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : 0.066692 a.u. 0.169513 Debye
Total : 0.066692 a.u. 0.169513 Debye
Total MCSCF time: 00:00:00
List of oscillator strengths greather than 1e-10
From to Energy (eV) Oscillator strength (length and velocity)
1 2 8.08798 8.153103e-03 3.791691e-02
List of rotatory strengths greather than 1e-10
From to Energy (eV) Rot. strength (a.u. and 10^-40 cgs)
Multi-Configurational Self-Consistent Field Driver
====================================================
Active space definition:
------------------------
Number of inactive (occupied) orbitals: 2
Number of active orbitals: 5
Number of virtual orbitals: 6
This is a CASSCF wavefunction: CAS(6,5)
CI expansion:
-------------
Number of determinants: 100
╭────────────────────────────────────╮
│ Driver settings │
╰────────────────────────────────────╯
Number of states : 3
State-averaged calculation
- Equal-weights
Max. iterations : 50
BFGS window : 5
Convergence thresholds:
- Energy change : 1e-08
- Gradient sq. norm : 1e-08
MCSCF Iterations
-------------------
Iter. | Average Energy | E. Change | Grad. Norm | CI Iter. | Trust rad. | Time
---------------------------------------------------------------------------------
1 -75.724068655 0.0e+00 1.2e-03 0 0.40 0:00:00
2 -75.725200119 -1.1e-03 9.4e-05 0 0.40 0:00:00
3 -75.725331151 -1.3e-04 3.2e-06 0 0.48 0:00:00
4 -75.725334435 -3.3e-06 3.2e-07 0 0.48 0:00:00
5 -75.725334997 -5.6e-07 3.8e-08 0 0.58 0:00:00
6 -75.725335073 -7.5e-08 8.7e-09 0 0.69 0:00:00
7 -75.725335090 -1.7e-08 2.2e-10 0 0.80 0:00:00
8 -75.725335090 -5.3e-10 3.7e-11 0 0.80 0:00:00
** Convergence reached in 8 iterations
9 -75.725335090 -3.5e-11 4.7e-12 0 0.80 0:00:00
Final results
-------------
* State 1
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -76.0098634104323
- Natural orbitals
1.98623 1.99931 1.97762 0.01412 0.02272
* State 2
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.71760513688933
- Natural orbitals
1.99620 1.00000 1.97731 1.00100 0.02548
* State 3
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -75.4485367237144
- Natural orbitals
1.99442 1.99771 1.00266 0.99771 0.00751
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 1:
--------------------------
Occupation: 2.000 Energy: -20.66135 a.u.
( 1 O 1s : 1.00)
Molecular Orbital No. 2:
--------------------------
Occupation: 2.000 Energy: -1.25118 a.u.
( 1 O 1s : -0.22) ( 1 O 2s : 0.52) ( 1 O 3s : 0.56)
( 1 O 1p0 : 0.18)
Molecular Orbital No. 3:
--------------------------
Occupation: 1.988 Energy: -0.84539 a.u.
( 1 O 1p0 : -0.56) ( 1 O 2p0 : -0.36) ( 2 H 1s : 0.19)
( 3 H 1s : 0.19)
Molecular Orbital No. 4:
--------------------------
Occupation: 1.666 Energy: -0.50141 a.u.
( 1 O 1p-1: -0.66) ( 1 O 2p-1: -0.48)
Molecular Orbital No. 5:
--------------------------
Occupation: 1.648 Energy: -0.70435 a.u.
( 1 O 1p+1: -0.55) ( 1 O 2p+1: -0.28) ( 2 H 1s : 0.25)
( 3 H 1s : -0.25)
Molecular Orbital No. 6:
--------------------------
Occupation: 0.675 Energy: 0.08480 a.u.
( 1 O 2s : -0.16) ( 1 O 3s : -1.07) ( 1 O 1p0 : 0.31)
( 1 O 2p0 : 0.39) ( 2 H 2s : 0.92) ( 3 H 2s : 0.92)
Molecular Orbital No. 7:
--------------------------
Occupation: 0.023 Energy: 0.55591 a.u.
( 1 O 1p+1: 0.77) ( 1 O 2p+1: 0.18) ( 2 H 1s : 0.44)
( 2 H 2s : 0.51) ( 3 H 1s : -0.44) ( 3 H 2s : -0.51)
Molecular Orbital No. 8:
--------------------------
Occupation: 0.000 Energy: 0.84550 a.u.
( 1 O 1p+1: -0.26) ( 1 O 2p+1: 0.40) ( 2 H 1s : -0.75)
( 2 H 2s : 1.44) ( 3 H 1s : 0.75) ( 3 H 2s : -1.44)
Molecular Orbital No. 9:
--------------------------
Occupation: 0.000 Energy: 1.08882 a.u.
( 1 O 1p-1: -0.95) ( 1 O 2p-1: 1.05)
Dipole moment for state 1
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.902880 a.u. -2.294892 Debye
Total : 0.902880 a.u. 2.294892 Debye
Dipole moment for state 2
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : 0.000000 a.u. 0.000000 Debye
Z : 0.071924 a.u. 0.182813 Debye
Total : 0.071924 a.u. 0.182813 Debye
Dipole moment for state 3
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.220125 a.u. -0.559501 Debye
Total : 0.220125 a.u. 0.559501 Debye
Total MCSCF time: 00:00:00
List of oscillator strengths greather than 1e-10
From to Energy (eV) Oscillator strength (length and velocity)
1 2 7.95275 8.899035e-03 4.003629e-02
1 3 15.27448 3.502994e-01 3.169096e-01
List of rotatory strengths greather than 1e-10
From to Energy (eV) Rot. strength (a.u. and 10^-40 cgs)
Multi-Configurational Self-Consistent Field Driver
====================================================
Active space definition:
------------------------
Number of inactive (occupied) orbitals: 2
Number of active orbitals: 5
Number of virtual orbitals: 6
This is a CASSCF wavefunction: CAS(6,5)
CI expansion:
-------------
Number of determinants: 100
╭────────────────────────────────────╮
│ Driver settings │
╰────────────────────────────────────╯
Number of states : 4
State-averaged calculation
- Equal-weights
Max. iterations : 50
BFGS window : 5
Convergence thresholds:
- Energy change : 1e-08
- Gradient sq. norm : 1e-08
MCSCF Iterations
-------------------
Iter. | Average Energy | E. Change | Grad. Norm | CI Iter. | Trust rad. | Time
---------------------------------------------------------------------------------
1 -75.650642982 0.0e+00 8.6e-03 0 0.40 0:00:00
2 -75.692220117 -4.2e-02 1.3e-03 0 0.40 0:00:00
3 -75.694350449 -2.1e-03 7.5e-05 0 0.48 0:00:00
4 -75.694439713 -8.9e-05 4.7e-06 0 0.48 0:00:00
5 -75.694445644 -5.9e-06 5.6e-07 0 0.48 0:00:00
6 -75.694446718 -1.1e-06 6.1e-08 0 0.58 0:00:00
7 -75.694446789 -7.2e-08 1.3e-08 0 0.58 0:00:00
8 -75.694446804 -1.5e-08 2.9e-10 0 0.69 0:00:00
9 -75.694446804 -2.8e-10 1.4e-11 0 0.69 0:00:00
** Convergence reached in 9 iterations
10 -75.694446804 -8.5e-12 1.8e-12 0 0.80 0:00:00
Final results
-------------
* State 1
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.99698121055859
- Natural orbitals
1.98702 1.98210 1.99942 0.01415 0.01731
* State 2
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.70831745311062
- Natural orbitals
1.99704 1.98460 1.00000 1.00024 0.01813
* State 3
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.62405336905377
- Natural orbitals
1.98743 1.99379 1.00000 0.02056 0.99822
* State 4
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -75.44843518488968
- Natural orbitals
1.99702 1.00111 1.99835 0.99825 0.00526
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 1:
--------------------------
Occupation: 2.000 Energy: -20.67802 a.u.
( 1 O 1s : -1.00)
Molecular Orbital No. 2:
--------------------------
Occupation: 2.000 Energy: -1.24525 a.u.
( 1 O 1s : 0.22) ( 1 O 2s : -0.52) ( 1 O 3s : -0.55)
( 1 O 1p0 : -0.20)
Molecular Orbital No. 3:
--------------------------
Occupation: 1.988 Energy: -0.87394 a.u.
( 1 O 1p0 : 0.56) ( 1 O 2p0 : 0.36) ( 2 H 1s : -0.18)
( 3 H 1s : -0.18)
Molecular Orbital No. 4:
--------------------------
Occupation: 1.734 Energy: -0.74018 a.u.
( 1 O 1p+1: -0.56) ( 1 O 2p+1: -0.28) ( 2 H 1s : 0.24)
( 3 H 1s : -0.24)
Molecular Orbital No. 5:
--------------------------
Occupation: 1.499 Energy: -0.46605 a.u.
( 1 O 1p-1: -0.67) ( 1 O 2p-1: -0.48)
Molecular Orbital No. 6:
--------------------------
Occupation: 0.512 Energy: 0.10271 a.u.
( 1 O 2s : 0.16) ( 1 O 3s : 1.07) ( 1 O 1p0 : -0.30)
( 1 O 2p0 : -0.39) ( 2 H 2s : -0.92) ( 3 H 2s : -0.92)
Molecular Orbital No. 7:
--------------------------
Occupation: 0.267 Energy: 0.23802 a.u.
( 1 O 1p+1: 0.45) ( 1 O 2p+1: 0.67) ( 2 H 1s : 0.18)
( 2 H 2s : 1.19) ( 3 H 1s : -0.18) ( 3 H 2s : -1.19)
Molecular Orbital No. 8:
--------------------------
Occupation: 0.000 Energy: 0.99722 a.u.
( 1 O 2p+1: -0.62) ( 2 H 1s : -0.97) ( 2 H 2s : 0.66)
( 3 H 1s : 0.97) ( 3 H 2s : -0.66)
Molecular Orbital No. 9:
--------------------------
Occupation: 0.000 Energy: 1.08042 a.u.
( 1 O 1p-1: 0.94) ( 1 O 2p-1: -1.05)
Dipole moment for state 1
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.865079 a.u. -2.198812 Debye
Total : 0.865079 a.u. 2.198812 Debye
Dipole moment for state 2
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : 0.000000 a.u. 0.000000 Debye
Z : 0.085985 a.u. 0.218553 Debye
Total : 0.085985 a.u. 0.218553 Debye
Dipole moment for state 3
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.042742 a.u. -0.108641 Debye
Total : 0.042742 a.u. 0.108641 Debye
Dipole moment for state 4
---------------------------
X : 0.000000 a.u. 0.000001 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.237138 a.u. -0.602745 Debye
Total : 0.237138 a.u. 0.602745 Debye
Total MCSCF time: 00:00:01
List of oscillator strengths greather than 1e-10
From to Energy (eV) Oscillator strength (length and velocity)
1 2 7.85494 7.812051e-03 4.149862e-02
1 4 14.92670 4.583994e-01 3.096522e-01
List of rotatory strengths greather than 1e-10
From to Energy (eV) Rot. strength (a.u. and 10^-40 cgs)
Multi-Configurational Self-Consistent Field Driver
====================================================
Active space definition:
------------------------
Number of inactive (occupied) orbitals: 2
Number of active orbitals: 5
Number of virtual orbitals: 6
This is a CASSCF wavefunction: CAS(6,5)
CI expansion:
-------------
Number of determinants: 100
╭────────────────────────────────────╮
│ Driver settings │
╰────────────────────────────────────╯
Number of states : 5
State-averaged calculation
- Equal-weights
Max. iterations : 50
BFGS window : 5
Convergence thresholds:
- Energy change : 1e-08
- Gradient sq. norm : 1e-08
MCSCF Iterations
-------------------
Iter. | Average Energy | E. Change | Grad. Norm | CI Iter. | Trust rad. | Time
---------------------------------------------------------------------------------
1 -75.630250704 0.0e+00 3.2e-03 0 0.40 0:00:00
2 -75.672532115 -4.2e-02 4.3e-03 0 0.40 0:00:00
3 -75.683260721 -1.1e-02 9.7e-04 0 0.56 0:00:00
4 -75.686039130 -2.8e-03 9.2e-06 0 0.67 0:00:00
5 -75.686051087 -1.2e-05 5.5e-07 0 0.67 0:00:00
6 -75.686051424 -3.4e-07 2.6e-08 0 0.67 0:00:00
7 -75.686051445 -2.1e-08 3.7e-10 0 0.80 0:00:00
8 -75.686051445 -2.8e-10 2.9e-11 0 0.80 0:00:00
** Convergence reached in 8 iterations
9 -75.686051445 -2.4e-11 1.5e-12 0 0.80 0:00:00
Final results
-------------
* State 1
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.98214396904912
- Natural orbitals
1.98461 1.99145 1.99958 0.01560 0.00875
* State 2
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -75.70439717085155
- Natural orbitals
1.98783 1.99722 1.00000 0.01582 0.99913
* State 3
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -75.62069022907518
- Natural orbitals
1.99499 1.99411 1.00000 0.99764 0.01326
* State 4
- S^2 : -0.00 (multiplicity = 1.0 )
- Energy : -75.60257216026119
- Natural orbitals
1.96066 1.22366 1.99968 0.04213 0.77386
* State 5
- S^2 : 0.00 (multiplicity = 1.0 )
- Energy : -75.52045369604085
- Natural orbitals
1.97487 1.01895 1.99956 0.97610 0.03052
Spin Restricted Orbitals
------------------------
Molecular Orbital No. 1:
--------------------------
Occupation: 2.000 Energy: -20.68941 a.u.
( 1 O 1s : -1.00)
Molecular Orbital No. 2:
--------------------------
Occupation: 2.000 Energy: -1.44678 a.u.
( 1 O 1s : 0.22) ( 1 O 2s : -0.49) ( 1 O 3s : -0.48)
Molecular Orbital No. 3:
--------------------------
Occupation: 1.976 Energy: -0.80534 a.u.
( 1 O 1p+1: -0.55) ( 1 O 2p+1: -0.28) ( 2 H 1s : 0.23)
( 3 H 1s : -0.23)
Molecular Orbital No. 4:
--------------------------
Occupation: 1.604 Energy: -0.58138 a.u.
( 1 O 2s : 0.18) ( 1 O 3s : 0.24) ( 1 O 1p0 : 0.60)
( 1 O 2p0 : 0.40)
Molecular Orbital No. 5:
--------------------------
Occupation: 1.600 Energy: -0.50270 a.u.
( 1 O 1p-1: -0.67) ( 1 O 2p-1: -0.48)
Molecular Orbital No. 6:
--------------------------
Occupation: 0.414 Energy: 0.21172 a.u.
( 1 O 1p+1: -0.43) ( 1 O 2p+1: -0.69) ( 2 H 2s : -1.24)
( 3 H 2s : 1.24)
Molecular Orbital No. 7:
--------------------------
Occupation: 0.406 Energy: 0.11950 a.u.
( 1 O 2s : 0.17) ( 1 O 3s : 1.09) ( 1 O 1p0 : -0.25)
( 1 O 2p0 : -0.41) ( 2 H 2s : -0.93) ( 3 H 2s : -0.93)
Molecular Orbital No. 8:
--------------------------
Occupation: 0.000 Energy: 1.00726 a.u.
( 1 O 2p+1: -0.69) ( 2 H 1s : -0.97) ( 2 H 2s : 0.59)
( 3 H 1s : 0.97) ( 3 H 2s : -0.59)
Molecular Orbital No. 9:
--------------------------
Occupation: 0.000 Energy: 1.07263 a.u.
( 1 O 1p-1: 0.94) ( 1 O 2p-1: -1.05)
Dipole moment for state 1
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : -0.847570 a.u. -2.154309 Debye
Total : 0.847570 a.u. 2.154309 Debye
Dipole moment for state 2
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : 0.000000 a.u. 0.000000 Debye
Z : 0.071706 a.u. 0.182257 Debye
Total : 0.071706 a.u. 0.182257 Debye
Dipole moment for state 3
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : 0.000000 a.u. 0.000000 Debye
Z : -0.023140 a.u. -0.058815 Debye
Total : 0.023140 a.u. 0.058815 Debye
Dipole moment for state 4
---------------------------
X : 0.000000 a.u. 0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : 0.110856 a.u. 0.281768 Debye
Total : 0.110856 a.u. 0.281768 Debye
Dipole moment for state 5
---------------------------
X : -0.000000 a.u. -0.000000 Debye
Y : -0.000000 a.u. -0.000000 Debye
Z : 0.135447 a.u. 0.344272 Debye
Total : 0.135447 a.u. 0.344272 Debye
Total MCSCF time: 00:00:01
List of oscillator strengths greather than 1e-10
From to Energy (eV) Oscillator strength (length and velocity)
1 2 7.55788 1.182600e-02 2.629917e-02
1 4 10.32868 1.101141e-01 1.302122e-01
1 5 12.56323 1.491871e-01 1.439096e-01
List of rotatory strengths greather than 1e-10
From to Energy (eV) Rot. strength (a.u. and 10^-40 cgs)
import matplotlib.pyplot as plt
import numpy as np
plt.figure(figsize=(6,4))
x = np.array(range(2,6))
plt.plot(x,np.array(first_exc_energy), label='First SA-CASSCF excitation energy')
plt.title('Deteriotation of state-averaged CASSCF excitation energies')
plt.ylabel("Energies (eV)")
plt.xlabel("Number of states")
plt.axhline(y=FCI_excitation_energies[0],label ='FCI',color='red')
plt.xticks(x)
plt.legend()
plt.tight_layout(); plt.show()
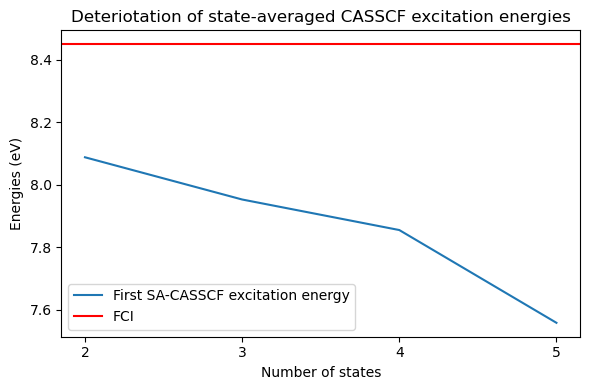
While for 2 states, the first excitation energy was relatively close to the fullCI, it degrades significantly as more states are added, with a difference of more than 0.5 eV going from 2 to 5 states. Adding dynamical correlation on-top of the CASSCF typically reduces this error, but it is still an undesirable behaviour.
Still, because of its simplicity and its reasonable accuracy, SA-MCSCF is one of the most frequently used technique to compute properties at the MCSCF level.